Mise à jour du 7 mai 2020
Avant-propos :
Dans la situation actuelle d’urgence à répondre à un problème de santé publique majeur, toutes les disciplines de la Biologie sont mises à contribution dans leur domaine d’expertise. La Physiologie est une discipline qui intègre tous les mécanismes moléculaires jusqu’au niveau de l’organisme entier et peut donc tenter de donner un sens à certaines observations qui ont été rapportées ou publiées.
En cette période de doutes, de questions, de vraies et fausses affirmations ou déclarations, la SPBI souhaite mettre à contribution ses membres experts dans leur domaine, afin d’aider à mieux appréhender les informations qui circulent en ce moment.
Le lecteur qui se penche rapidement sur la problématique « système rénine-angiotensine (SRA) et COVID-19 » verra soulevée la question de l’arrêt des bloqueurs du SRA, ces derniers prédisposant potentiellement à l’infection ou à des formes plus sévères chez les patients contaminés, avec en effet des essais cliniques qui testent leur interruption, puis verra au contraire des recommandations de différentes sociétés savantes indiquant de ne pas modifier nos pratiques, ou enfin lira au contraire que ces traitements pourraient être protecteurs au cours du COVID-19 avec des essais thérapeutiques en cours qui testent leur efficacité…
Nous n’avons pas la prétention de pouvoir répondre à l’immensité des questions que pose la complexité de cette épidémie, mais d’assembler les faits disponibles s’agissant du lien entre COVID-19 et SRA, et de les replacer dans le contexte des connaissances actuelles dans le domaine, afin de clarifier le rôle que pourraient avoir le SRA et les traitements pharmacologiques modulateurs de cette voie dans les différentes phases de l’infection, et plus largement de discuter les problématiques qui impliquent le SRA au cours du COVID-19.
Le sujet fait l’objet de publications quotidiennes ces derniers jours (voir figure ci-dessous) et les connaissances peuvent donc évoluer très rapidement…mais voici quelques éléments en date du 7 mai 2020

Introduction :
COVID-19 et enzyme de conversion de l’angiotensine de type 2
Le mécanisme de l’infection par ce nouveau coronavirus, et en particulier les facteurs qui prédisposent à une forme sévère de COVID-19, restent encore largement incompris.
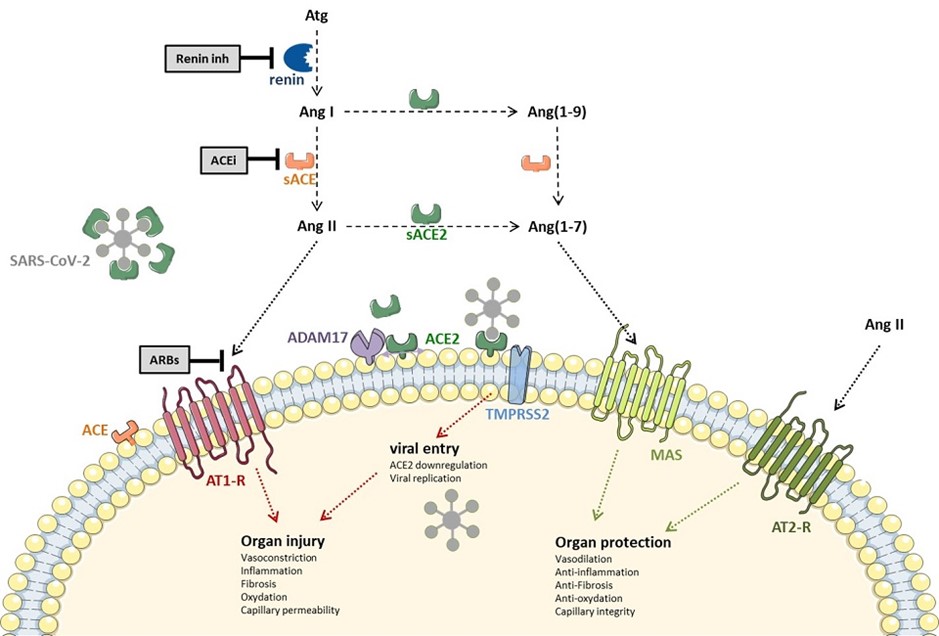
Le severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) a pour récepteur l’enzyme de conversion de l’angiotensine de type 2 (ACE2) membranaire (Hoffman, Cell, 2020 – Chen, BBRC, 2020), de même que la SARS-CoV(-1) (Li et al, Nature 2003, Kuba et al, Nature Medicine 2005). L’affinité du SARS-CoV-2 pour l’ACE2 semble cependant nettement supérieure à celle du SARS-CoV (Wrapp et al, Science 2020 ; 367 :1260-63).
L’ACE2 est une carboxypeptidase découverte au début des années 2000. Il s’agit d’un homologue de la beaucoup plus célèbre ACE (ou ACE1). Sur le plan physiologique, l’ACE2 est considérée comme une voie de modulation des effets de l’ACE, avec des effets non seulement cardiovasculaires, mais en fait bien au-delà, globalement « protecteurs » car favorisant la vasodilatation et limitant la fibrose.
ACE2 est exprimée dans l’épithélium pulmonaire comme l’ACE, mais aussi, entre autres, dans le système nerveux central, le cœur, les vaisseaux, le rein, et le testicule (Harmer, FEBS lett 532, 107110, 2002 ; Hamming et al, J Pathol 2004 ; 203 :631-37). Elle est également fortement exprimée dans l’intestin où elle joue un rôle important pour l’absorption de certains acides aminés, fait intéressant quand on sait la prévalence de l’atteinte digestive dans le COVID-19. Une étude conduite chez le lapin a mesuré l’expression d’ACE2 au cours de l’âge avec une comparaison males/femelles (Xuding et al, Life Sci 2006 ; 78 :2166-71). Les niveaux d’ACE2 diminuaient drastiquement avec l’âge et étaient plus bas chez les males âgés que les femelles âgées. En revanche, des études post-mortem d’expression pulmonaire de l’ACE2 humaine (Genotype-Tissue Expression project) ont montré une expression faible et similaire chez les femmes et les hommes.
L’ACE2 a pour substrat (non spécifique) un peptide composé de 8 acides aminés, l’angiotensine II (Ang II), qu’elle transforme en angiotensine (1-7) [Ang(1-7)]. Elle permet aussi la transformation de l’angiotensine I (Ang I) en Ang(1-7), via la synthèse d’un métabolite intermédiaire, l’angiotensine (19) [Ang(1-9)] (Donogue et al, Circ Res 2000 ; 87 :E1-9). Ang(1-7) agit via le récepteur MAS (appartenant à la famille des récepteurs couplés aux protéines G) avec des effets biologiques opposés à ceux de l’Ang II (notamment des effets vasodilatateurs, anti-inflammatoires, antiprolifératifs, protecteurs de la barrière capillaire, et anti-fibrosants).
Fait important, l’ACE2 n’est pas bloquée par les IEC (Tipnis et al, J Biol Chem 2000 ; 275 :33238-43), son site actif étant plus petit que celui de l’ACE.
Des communications récentes du Lancet Respir Med (Fang et al, Lancet Respir med 2020) et du BMJ (Sommerstein and Gräni, BMJ 2020), suivies de plusieurs autres (Esler and Elser J Hypertens 2020 ;38 :781-2 ; Diaz J Travel Med 2020 March18, Aronson and Ferner, BMJ April 2, 2020), largement relayées sur le net et les réseaux sociaux, ont soulevé l’hypothèse que les inhibiteurs de l’enzyme de conversion (IEC) et/ou antagonistes des récepteurs de l’angiotensine II (ARA2, ou « sartans ») pourraient augmenter l’expression d’ACE2 à la surface membranaire et ainsi constituer un facteur de vulnérabilité au COVID-19… De là à suggérer l’arrêt de ces traitements le temps de l’épidémie, il n’y a qu’un pas que certains avaient suggéré de franchir…
→ Voici quelques éléments de réflexion (forcément évolutifs !) concernant les discussions actuelles autour du lien entre ACE2 et COVID-19.
Pathologies cardiovasculaires/bloqueurs du SRA et prédisposition à la maladie COVID-19 ou à ses formes sévères
Comorbidités cardiovasculaires et COVID-19
Une donnée épidémiologique qui a initié la recherche d’un lien entre bloqueurs du système rénine angiotensine (SRA) et COVID-19 est la forte prévalence des pathologies cardiovasculaires et notamment de l’hypertension artérielle (HTA) chez les patients atteints du COVID-19, et plus particulièrement des formes sévères de la pathologie.
Il a été montré dans plusieurs cohortes que la prévalence de l’HTA est élevée chez les patients atteints du COVID-19, et qu’elle l’est plus encore chez les patients qui décèdent de la maladie. Le problème commun à toutes ces études est qu’il s’agit d’analyses non ajustées, en particulier sur l’âge qui est le premier facteur associé à la sévérité de la maladie, mais également sur l’indice de masse corporelle.
Dans une cohorte de 419 patients décédés du COVID-19 en Italie (ministère de Santé Italien, données du 20 mars, https://www.epicentro.iss.it/coronavirus/sars-cov-2-decessi-italia), 74% souffraient d’hypertension, 34% de diabète et 30% de cardiopathie ischémique. Dans cette même cohorte, 36% étaient préalablement traités par IEC et 16% par ARA2.
Plus récemment, une large série italienne a été publiée (Grasselli et al, JAMA April 6 2020). Chez 1591 patients admis en réanimation, 49% étaient hypertendus (alors que seulement 4% avaient un antécédent de BPCO), avec sans surprise une prévalence de l’HTA qui augmentait avec l’âge, de 11% chez les moins de 40 ans, à 75% chez les plus de 80 ans. 63 % des patients décédés étaient hypertendus versus 40% des patients qui sont sortis vivants de réanimation, mais les hypertendus étant plus âgés il est là-encore difficile d’en déduire un lien causal entre HTA et infection sévère.
En résumé, comme souligné par de nombreux auteurs, ces prévalences de pathologies cardiovasculaires semblent être celles attendues compte-tenu de l’âge des patients et des populations concernées (Tocci et al, J Hum Hypertens 2017;31:258–262 ; Sanchis-Gomar, Mayo Clin Proc 2020 in press). La prévalence de l’HTA est très élevée au-delà de 70 ans donc attendue très élevée chez les patients qui présentent les formes plus graves et a fortiori décèdent du COVID-19.
Traitement par bloqueurs du SRA et COVID-19
Assez rapidement a été soulevée l’hypothèse que le lien entre pathologies cardiovasculaires et COVID-19 puisse provenir de la prescription fréquente de bloqueurs du SRA chez ces patients, le mécanisme sous-jacent invoqué étant la surexpression de l’ACE2 sous ces traitements, voir plus bas.
Deux études chinoises rétrospectives et monocentriques (une rapportant 126 patients hypertendus traités, dont 43 sous IEC ou ARA2 à l’admission [Yang et al, medRxiv preprint https://doi.org/10.1101/2020.03.31.20038935], et une autre rapportant 42 patients hypertendus traités, dont 17 sous IEC ou ARA2 à l’entrée [Meng et al, Emerg Microbes Infect, 9: 757-760]) suggéraient que les IEC/ARA2 puissent être associés à un meilleur pronostic au cours de l’infection à COVID-19 (tendance non significative à moins de formes sévères en analyse non ajustée). Une étude multicentrique rapportant 511 patients dont 78 traités pour hypertension (et dont on peut penser qu’elle inclut les données de Meng et al, Emerg Microbes Infect, 9: 757-760) n’a mis en évidence aucune différence dans la sévérité de la maladie en fonction des différentes classes d’antihypertenseurs et en comparaison aux patients ne prenant aucun traitement antihypertenseur. Une sous-analyse de 46 patients âgés de plus de 65 ans montrait que la prise d’ARA2 semblait protectrice mais là-encore les limites méthodologiques sont majeures [Liu et al, medRxiv preprint https://doi.org/10.1101/2020.03.20.20039586]. Dans les trois études, il est difficile de comprendre si les analyses ont été effectuées en fonctions des traitements à l’entrée ou des traitements maintenus pendant le séjour.
Une étude monocentrique conduite dans l’hôpital central de Wuhan a rapporté les données de 1178 patients infectés par le COVID-19, dont 362 hypertendus (prévalence 30,7%) (Li et al, JAMA cardiology, published online April 23rd). Parmi les hypertendus, 115 (31,8%) prenaient des IEC ou ARA2 (prise du traitement définie par la prise sur la liste des traitements à l’entrée, poursuivie pendant le séjour). La mortalité des patients hypertendus était de 21,3%. Le pourcentage de patients prenant des IEC/ARA ne différait pas entre les morts et les survivants, ni entre les patients ayant fait une forme sévère ou non de l’infection. L’analyse séparée des IEC et des sartans donnait les mêmes résultats.
Une dernière étude chinoise, multicentrique rétrospective, de méthodologie plus détaillée et incluant des ajustements (Zhang et al, circulation res 2020) a analysé les données de 1128 patients hypertendus et atteints du COVID-19 (parmi une cohorte de 3430 patients, soit une prévalence d’hypertension de 33%), dont 188 étaient traités par IEC ou sartans pendant l’hospitalisation, et 940 ne recevaient aucun de ces traitements. Les taux de mortalités étaient de 3,7 et 9,8% dans les groupes avec et sans bloqueurs du SRA, respectivement (p=0.01). Ces patients ont été comparés par plusieurs approches. Dans un modèle de Cox ajusté sur âge, sexe, comorbidités et traitements reçus pendant le séjour, le risque de mortalité était plus faible dans le groupe bloqueurs du SRA (HR 0.42, 95% CI 0.19-0.92). Par une approche de score de propension avec ajustement supplémentaire des variables non équilibrées dans un modèle de cox, le HR était de 0.37 (0.15-0.89). Dans une analyse en sous-groupe avec matching par score de propension, le HR ajusté était de 0.30 (0.12-0.70). L’effet bénéfique sur la mortalité associé à la prise des bloqueurs du SRA pouvait traduire un biais résiduel, le traitement étant potentiellement maintenu chez les patients les moins sévères, mais les auteurs concluaient qu’a contrario leurs résultats rendaient improbable un sur-risque associé à ces traitements…
Une étude britannique (King’s College Hospital and Princess Royal University Hospital, Londres) conduite chez 205 patients hospitalisés avec un diagnostic de COVID-19 est actuellement en « preprint » (Bean et al, MedTXiv 2020 doi.org/10.1101/2020.04.07.20056788). La prise d’un IEC était définie par la prise de ces traitements pendant les 7 jours précédant l’admission ou pendant l’hospitalisation. Les patients dont le traitement était suspendu pendant l’hospitalisation était analysés dans le groupe « IEC ». Seuls 9 patients étaient sous sartans et ont été regroupés dans les « non-IEC ». L’analyse par régression logistique était brute, puis ajustée sur l’âge et le sexe, puis ajustée sur l’hypertension, et enfin sur les comorbidités autres (diabète, cardiopathie ischémique, insuffisance cardiaque). 37 patients ont ainsi été classés dans le groupe «IEC », et avaient un risque moindre de faire une forme sévère de COVID-19, jugé sur le critère composite mortalité ou admission en réanimation dans les 7 jours suivant le début des symptômes.
Une autre étude également en « pre-print » s’appuie sur la cohorte Veterans Affairs Birth (Rentsch et al, MedRxiv, doi.org/10.1101/2020.04.09.20059964). Chez 3789 vétérans âgés de 54 à 75 ans et testés pour le COVID-19 (dont 585 positifs), la prescription d’IEC ou ARA2 dans l’année qui précédait l’hospitalisation était soigneusement vérifiée, mais aucune donnée n’était disponible sur le maintien ou non de traitements pendant l’hospitalisation. 297 patients ont été hospitalisés, dont 122 en soins intensifs. L’exposition antérieure aux IEC ou ARA2 n’était pas associée au fait d’avoir une PCR positive. En analyse univariée, ces traitements étaient associés à une incidence plus élevée d’hospitalisation et de transfert aux soins intensifs, effet qui s’atténuait et n’était plus significatif après ajustement.
Ces quelques données préliminaires, toutes soumise à des biais importants, ne sont en tout état de cause, dans l’ensemble, pas en faveur d’un rôle délétère majeur de ces traitements.
Très récemment, le 1er mai, la parution en ligne, simultanément, de trois publications sur le sujet (Mancia et a, NEJM 2020 May 1st ; Mehra et al, NEJM 2020 May 1st ; Reynolds et al, NEJM 2020 May 1st), accompagnées d’un éditorial (Jarcho et al, NEJM 2020 May 1st) , dans le N Eng J Med sont venues quasiment clore le débat.
L’étude conduite par Mancia et al a rapporté les données de 6272 patients avec infection à COVID-19 confirmée en Lombardie (Italie) comparées à celles de 307759 contrôlés appariés pour âge, sexe et ville de résidence. Une analyse de régression logistique multivariée conditionnelle n’a retrouvé aucune association entre la prise d’ARA2 ou IEC et la survenue d’une infection par COVID-19 d’une part, et avec la survenue de formes sévères de la maladie d’autre part.
Mahra et al ont conduit une étude dans 11 pays chez 8910 patients ayant une infection à COVID-19 et hospitalisés. Dans une analyse de régression logistique multivariée, ni les IEC ni les sartans n’étaient associés à la mortalité intra-hospitalière.
Enfin, l’étude de Reynolds et al a été conduite à partir des données de 12594 patients testés pour le COVID-19 à New-York (New York University Langone Health electronic health record). Parmi 5894 patients testés positifs, 1002 ont développé une forme sévère (définie par l’admission en réanimation, le recours à la ventilation mécanique ou le décès). La prise des traitements était obtenue par interrogation de la base de la donnée de santé, et définie par une prescription dans les 18 mois qui ont précédé et en l’absence d’argument en faveur d’une interruption dans le mois qui a précédé le test. Une analyse a été conduite à la fois dans l’ensemble de la population et chez les hypertendus (études de la prédisposition à un test positif), ainsi que dans le sous-groupe des patients COVID-19, à nouveau tous patients puis patients hypertendus (analyse de la prédisposition à une forme sévère). Une analyse bayésienne n’a pas retrouvé d’association positive avec la prise de bloqueurs du SRA (ni avec aucun autre anti-hypertenseur), que ce soit pour un test positif ou une forme sévère chez les patients positifs, après appariement avec un score de propension pour chaque classe thérapeutique.
Cette dernière étude cherchait donc à définir, comme celle de Rentsch et al (MedRxiv, doi.org/10.1101/2020.04.09.20059964) citée plus haut, non seulement si la prise de bloqueurs du SRA prédisposait à faire une forme sévère de la pathologie, mais également si ces traitements prédisposaient à faire une infection (que cette dernière soit sévère ou non le cas échéant), par comparaison de patients testés positifs et négatifs.
Une troisième étude parue en ligne le 5 mai (Mehta et al, JAMA cardiology 2020) avait également ce double objectif et se prononce spécifiquement sur la question du risque d’un test positif associé aux bloqueurs du SRA. Il s’agit encore d’une étude rétrospective, à partir de données de Cleveland Clinic Health System obtenues en Ohio et en Floride aux USA. La population était composée de 18472 patients testés pour le COVID-19, dont 12,4% prenaient un bloqueur du SRA (d’après la donnée renseignée au moment du test, sans information sur la poursuite du traitement au décours). 1735 patients (9,4%) ont eu un test positif, et parmi eux 421 (24,3%) ont été hospitalisés, dont 161 admis en réanimation, et parmi eux 111 placés sous ventilation mécanique. Les auteurs ne mettent en évidence aucune association entre la prise d’un bloqueur du SRA et le risque d’avoir un test COVID19 positif chez les 18472 patients testés (odds ratio pondéré par score de propension 0.97 [95%CI 0.81-1.15]). En analyse secondaire, les auteurs ont étudié l’association entre la prise d’un bloqueur du SRA et les critères de jugement cliniques. Le HR pondéré pour le critère hospitalisation était de 1.93 (95%CI 1.38-2.71), celui du critère admission en réanimation était de 1.64 (95% CI 1.07-2.51), et celui du critère ventilation mécanique de 1.32 (95% CI 0.80-2.18). Si l’on regarde séparément IEC et ARA, le sur-risque d’admission en réanimation n’était pas significatif pour les ARA2. Les auteurs restent très prudents sur l’interprétation de ce sur-risque, et évoquent avant tout la possibilité d’un biais lié aux affections cardiovasculaires sous-jacentes, insuffisamment pris en compte par le score de propension.
Au total, aucune donnée épidémiologique solide ne permet de se prononcer sur le fait que les bloqueurs du SRA prédisposeraient à faire une infection COVID-19 ou à faire une forme grave de la pathologie. A contrario, les trois publications du NEJM parues le 1er mai, sur de grandes populations, dans des pays différents, et avec des méthodologies différentes, bien qu’observationnelles, sont un argument très fort pour penser que la prise au long cours d’IEC ou ARA2 ne prédispose ni à faire des infections par le SRAS-CoV-2, ni à développer des formes plus sévères de la maladie.
Les données concernant la poursuite ou non des traitements au cours de l’infection avérée restent parcellaires et il est plus difficile de se prononcer sur ce point. On peut bien entendu émettre l’hypothèse qu’une prise au long cours de bloqueurs du SRA préalable à l’admission constitue un biais prédisposant à une maladie plus sévère (patients hypertendus, atteints de pathologies cardionéphrologiques), mais a contrario le maintien de ces traitements pendant l’hospitalisation pourrait être le signe d’une affection moins sévère (car le traitement est plus souvent maintenu chez les patients qui n’ont pas de de complication rénale, elle-même facteur de mauvais pronostic, comme montré par Cheng et al ; Kidney Int 2020 ; 97 :829-38…). Il faut donc des études de grande taille permettant de tenir compte le plus finement possible des facteurs confondants, et avec une documentation précise s’agissant de la prescription des traitements et de leur éventuelle interruption, pour répondre à ces questions de la manière la plus fiable possible dans des études observationnelles.
Deux essais thérapeutiques vont tester de manière randomisée l’arrêt des bloqueurs du SRA chez les patients qui en prenaient au long cours et qui présentent une infection par le COVID-19 : un en France, ACORES-2, ClinicalTrials.gov Identifier: NCT04329195, et un en Irlande, CORONACION, NCT04330300.
Nous n’avons pas connaissance d’essai qui teste l’effet de l’arrêt des traitements bloqueurs du SRA en préventif, de fait de telles études seraient méthodologiquement plus compliquées et surtout discutables au vu des résultats sus-cités.
Effet supposé/attendu des IEC et sartans sur ACE2
Nous allons voir plus en détail sur quel rationnel repose le lien suspecté en bloqueurs du SRA et COVID-19. Les données disponibles concernant l’effet du blocage pharmacologique du SRA sur ACE2 ne sont pas uniformes.
Données chez l’animal
Une étude publiée en 2005 (Ferrario et al, circulation 2005 ;111:2605-2610) sur un modèle de rat montre que lisinopril et losartan augmentent l’expression de l’ARNm de l’ACE2 dans le cœur, ainsi que l’activité de l’enzyme. Cette étude a été largement citée dans les controverses récentes sur le sujet. Dans le rein en revanche, la même équipe a montré que l’ARNm de l’ACE2 n’était modifié ni sous lisinopril ni sous losartan, alors que l’activité de l’enzyme était augmentée sous les deux traitements (Ferrario et al Kidney Int 2005 ; 68 : 2189–2196). Plusieurs autres études chez l’animal ont retrouvé des résultats contradictoires, certaines confirmant une augmentation de l’ARNm et/ou de la protéine et/ou de l’activité de l’ACE2 (notamment dans le cœur) sous IEC et/ou ARA2 (exemple Soler et al, AJP Renal Physiology 2009) tandis que d’autres ne montraient aucune modification de ces mêmes marqueurs (exemple Burchill et al, Clin Sci (Lond) 2012). Pour revue de la modulation pharmacologique de l’ACE2 sur modèles animaux, voir Soler et al, Curr Hypertens Rep 2008, 10: 410 – 414.
Données chez l’homme
Chez l’homme, les données sont également contradictoires, et surtout très parcellaires. Une étude observationnelle (Furahashi, Am J Hypertens 2015 ; 28 :15-21) conduite chez 100 hypertendus traités et 100 sujets contrôles non traités a analysé le niveau d’ACE2 urinaire chez des patients traités par enalapril, amlodipine, ou plusieurs sartans (losartan, candésartan, valsartan, telmisartan, et olmesartan). Le seul traitement associé à une augmentation de l’ACE2 urinaire était l’olmesartan.
Chez 24 patients ayant une HTA essentielle, l’administration aigue de captopril (un IEC) n’avait aucun effet sur la production d’Ang(1-7), mais cette dernière augmentait après 6 mois de traitement (Luque et al, J Hypertens. 1996;14:799-805). Cependant plusieurs autres enzymes peuvent aboutir à la synthèse d’Ang(1-7) (Serfozo, Hypertension 2020;75:173-182) et l’activité tissulaire d’ACE2 n’était pas directement évaluée.
Chez 79 coronariens (Ramchand et al, PlosOne2018), le niveau d’ACE2 circulante était associé à la survenue ultérieure d’évènements cardiovasculaires, mais ne montrait aucune corrélation avec la prise d’un bloqueur du SRA. Fait notable, les hommes avaient une activité ACE2 supérieure à celle des femmes.
En tout état de cause, aucune donnée n’est disponible à notre connaissance concernant l’expression pulmonaire d’ACE2 sous bloqueurs du SRA.
Synthèse
En résumé, si en effet la voie de synthèse d’Ang(1-7) est probablement activée lors du blocage du SRA via les IEC et/ou les ARA2, l’effet sur l’expression protéique du récepteur ACE2 à la surface membranaire (donc celui qui nous intéresse dans le contexte actuel) n’est pas clairement établi, et encore moins dans le poumon, ce d’autant qu’il semble être très différent en fonction des tissus et des espèces. L’absence de parallélisme entre l’ARNm et la protéine a été bien montré dans plusieurs situations en tout cas, et l’expression d’ARNm (argument évoqué lors des controverses sus-citées sur la base des études de Ferrario, Circulation, 2005) ne saurait présager de l’augmentation du niveau des protéines à la surface membranaire dans les différents organes (poumon, cœur, rein, intestin…).
Une excellente synthèse de l’ensemble des études ayant analysé l’effet des bloqueurs du SRA sur l’ARNm et/ou protéine (activité/niveau d’expression) a été publiée dans la revue Cardiovascular research le 15 avril (Kreutz et al, Cardiovasc Res 2020 Apr 15 doi: 10.1093/cvr/cvaa097).
ARA2 versus IEC
Existe-t-il un effet différentiel « sartans/IEC » ? : Cet aspect peut se discuter. Cela dépend du/des mécanisme(s) par le/lesquels les voies ACE-AngII et ACE2-Ang(1-7) se modulent mutuellement. Est-ce que l’augmentation d’Ang II est nécessaire et suffisante à activer ACE2, ou bien est-ce que l’augmentation d’Ang I y contribue, ou le mécanisme est-il encore différent ? Ferrario et al (cf. les deux publications citées ci-dessus, datant de 2005) expliquent que, dès lors que le récepteur AT1 est moins activé (soit parce que bloqué par un sartan soit parce qu’en amont un IEC bloque la formation d’Ang II), alors ACE2 est activée. Selon ces auteurs, tout passe par le récepteur AT1 et donc IEC et sartans ont des effets attendus relativement similaires. Selon Deshotel et al (Hypertension 2014) l’Ang II conduit à l’ubiquitination et donc la dégradation de l’ACE2 membranaire en se liant à son récepteur AT1 (ce qui modifierait les interactions à la membrane des deux récepteurs). Les sartans ou ARA2 pourraient donc jouer un rôle modulateur important dans ce mécanisme, mais cela reste spéculatif.
Expression d’ACE2 et pathologies cardiovasculaires et respiratoires
Indépendamment des effets de la modulation pharmacologique de la voie de l’Ang II sur ACE2, il est à noter que les pathologies comme le diabète, l’HTA et l’insuffisance cardiaque sont associées à une augmentation de l’expression de l’ACE2, ou tout du moins de l’enzyme circulante. Les données sont plus complexes sur les modèles animaux car tout dépend de la place de la voie ACE2/Ang(1-7) dans le modèle (cause ou conséquence de la pathologie étudiée). L’ACE2 est le plus souvent diminuée dans les modèles expérimentaux d’hypertension (Esler and Elser J Hypertens 2020 ;38 :781-2).
Quant à l’expression pulmonaire de l’ACE2, elle est possiblement fortement modifiée dans diverses pathologies (BPCO…), et ce dans des ordres de grandeur bien plus significatifs que celles que pourraient entrainer un blocage chronique du SRA, mais ces hypothèses restent à vérifier.
Au cours des pathologies cardiovasculaires et notamment dans l’insuffisance cardiaque, l’augmentation des concentrations circulantes d’ACE2 est en outre un facteur pronostique péjoratif (Epelman, J Card Fail 2009).
Dans une réponse au commentaire sus-cité paru dans lancet Respir Med (Fang et al, Lancet Respir med 2020), Tignanelli et al suggèrent qu’une hyperactivation spontanée de la voie ACE-Ang II est une hypothèse tout aussi plausible qu’une surexpression de l’ACE2 pour expliquer la susceptibilité des patients hypertendus au COVID-19 (Tignanelli et al, Lancet Respir Med 2020, March 26).
Expression membranaire d’ACE2 et passage intracellulaire du virus ?
Par ailleurs, le récepteur ACE2 seul ne permet pas le passage du SARS-CoV2 dans la cellule. Le virus se fixe à sa cellule cible en se liant au récepteur ACE2 par l’intermédiaire de la sous-unité S1 de la protéine S (la protéine du Spike). La pénétration intracellulaire du virus nécessite une étape supplémentaire, à savoir le clivage ou priming de la protéine S par des protéases intracellulaires, ce qui libère alors un site de la sous-unité S2 responsable de la fusion du virus avec la membrane cellulaire. La sérine protéase TMPRSS2 est responsable de ce priming s’agissant du SARS-CoV-2 et sa coexpression par la cellule est indispensable à la pénétration intracellulaire du virus (Hoffmann et al, Cell, 2020). Notons à cet effet que le camistat mesylate est un bloqueur de TMPRSS2, mais à ce stade, nous n’avons pas connaissance d’essais en cours avec ce composé dans le COVID-19.
Dans des séries autopsiques de patients décédés du SARS (infection par le SARS-CoV, qui lui aussi a pour récepteur membranaire ACE2), le virus a été retrouvé dans des cellules ACE2-négatives tandis que des cellules ACE2-positives restaient indemnes (Gu and Korteweg, Am J Pathol 2007). Donc ACE2 ne serait donc ni indispensable, ni suffisant pour qu’une cellule soit infectée par le virus.
Le lien entre le niveau d’expression d’ACE2 – potentiellement modifié par les bloqueurs du SRA – et la pénétration intracellulaire du virus reste donc totalement hypothétique. Certains suggèrent en effet que l’augmentation de l’expression d’ACE2 membranaire par les bloqueurs du SRA ne soit pas accompagnée d’une augmentation parallèle de TMPRSS2, essentielle à la pénétration intracellulaire du virus. Selon cette hypothèse, l’augmentation d’ACE2 permettrait de « neutraliser » les virus à la surface cellulaire…Une hypothèse voisine suggère que les ARA2 pourraient stabiliser les complexes AT1R-ACE2 et empêcher le virus d’interagir avec son récepteur…
→ Autant de pistes à explorer qui montrent que le rôle délétère de l’activation de l’ACE2 – potentiellement induite par les bloqueurs du SRA – n’est qu’une hypothèse parmi tant d’autres et doit être traitée comme telle à ce jour.
Dangers associés à un arrêt des bloqueurs du SRA
En revanche, on peut aisément anticiper les effets dévastateurs d’un arrêt de ces traitements cardionéphroprotecteurs au niveau mondial, avec notamment un pourcentage incertain de reprise de ces traitements au décours de l’épidémie. Non seulement le bénéfice de ces thérapeutiques est largement prouvé (Vaduganathan et al, N Eng J Med, March30 2020), mais la dangerosité de leur arrêt, dès les premières semaines, (au moins dans l’insuffisance cardiaque) l’est également (exemple Halliday et al, TRED-HF, Lancet 2019;393:61–73.).
Qu’en est-il des recommandations des sociétés savantes cliniques concernant l’usage des IEC/Sartan en cas d’infection par le COVID ?
L’ensemble de ces données explique les avis de la plupart des sociétés savantes (voir tableau récapitulatif ci-dessous) qui s’accordent à dire que rien n’incite à arrêter en préventif les IEC ou sartans (ARA2) dans la population.
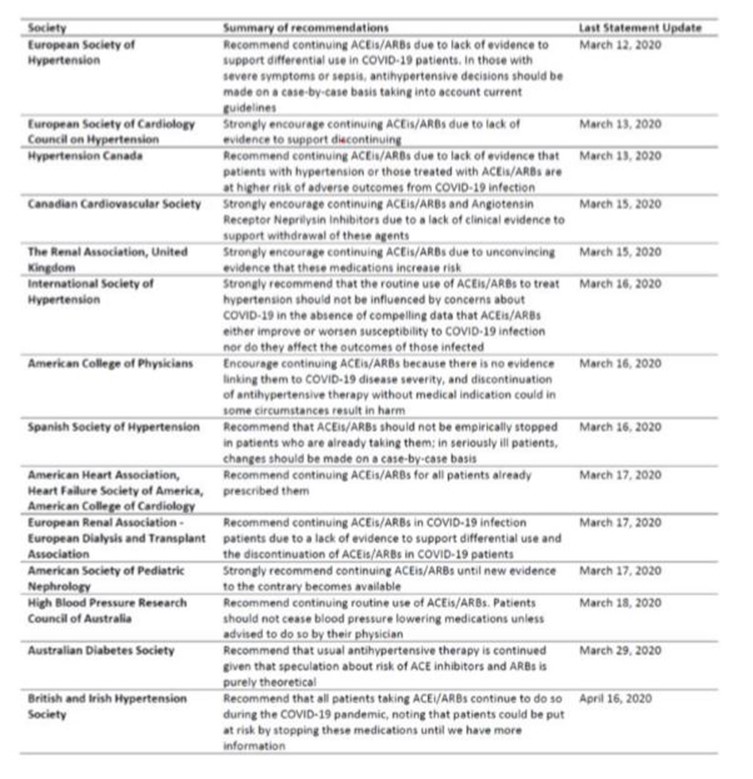
Pour revue détaillée des recommandations des sociétés savantes, voir également le viewpoint publié le 3 avril dans JAMA cardiology (Bavishi et al, JAMA cardio April 3 2020).
Une hypothèse inverse : équilibre AngII/Ang(1-7) et dommages d’organes
A contrario, on peut émettre l’hypothèse que les voies globalement « protectrices », pulmonaire, cardiovasculaire et rénale médiées par l’Ang(1-7), soient partiellement inhibées par la liaison du virus à l’enzyme ACE2. Le cas échéant, la balance homéostatique ACE-Ang II/ACE2-Ang(1-7) est déviée vers la voie ACE-Ang II, et ne peut qu’être alors encore plus défavorablement influencée par l’arrêt d’un bloqueur du SRA… Est-ce que cette inhibition fonctionnelle de l’ACE2 par le virus participe à la pathogénie et à l’immuno-modulation au cours du COVID-19 ? Voilà une piste qui fait couler beaucoup d’encre.
ACE2 et atteinte pulmonaire : données chez l’animal
Le rôle protecteur de la voie ACE2-Ang(1-7) et a contrario délétère de la voie ACE-AngII a été montré dans différents modèles murins d’infection pulmonaire (Imai et al, Nature 2005 ; 43 :112-116 ; Kuba, Nat Med 2005 ; 11 :875-79 ; Wosten-van Asperen, J Pathol 2011 ; 225 :618-27 ; Yang et al Sci Rep. 2014; 4: 7027, Gu et al, Sci Rep 2016 ; 6 :19840 ; Yan et al, 2015 Sci China Life Sci ; 58(2):208-11 ; Zou et al, Nat Commun 2014;5:3594).
Dans plusieurs modèles de lésion pulmonaire aigue non liées au SARS (inhalation de liquide gastrique, sepsis…), Imai et al (Nature 2005 ; 43 :112-116 ) ont démontré le rôle protecteur de la voie ACE2. Ainsi, des souris KO ACE2 avaient des lésions pulmonaires plus sévères que des souris wildtype. En outre, l’expression de la protéine ACE2 était drastiquement diminuée dans le poumon des souris wild-type infectées, le niveau d’Ang II augmentait dans le poumon et le plasma, et l’ACE2 recombinante protégeait des atteintes pulmonaires sévères, cet effet protecteur n’étant observé que si le site catalytique de l’ACE2 recombinante était intact. A contrario, les mêmes auteurs montrent que les souris ACE-/- KO sont partiellement protégées des lésions pulmonaires induites par l’inhalation de liquide gastrique. Un effet protecteur intermédiaire était observé chez les souris KO ACE+/-. En outre, la délétion homozygote pour le gène de l’ACE corrigeait le phénotype très sévère observé chez les souris KO ACE2 dans les différents modèles d’agression pulmonaire. Enfin, les auteurs ont démontré que le rôle délétère de la voie ACE-AngII est médié par les récepteurs AT1 de l’Ang II. D’une part, les souris KO pour ce récepteur ont de moindres lésions pulmonaires (l’inverse étant observé en cas d’inactivation des récepteurs AT2), et d’autre part, l’inhibition pharmacologique des récepteurs AT1 atténuait la sévérité des lésions pulmonaires chez les souris KO ACE2.
L’infection pulmonaire par les virus du SARS présente des particularités liées au rôle d’acteur pathogène d’ACE2 dans cette affection. Dans une étude complémentaire de modèle murin de lésion pulmonaire par infection par le virus du SARS-CoV, la même équipe a montré que les souris KO ACE2 sont protégées de l’infection en comparaison aux souris sauvages (atténuation très marquée de la réplication virale et moindres lésions pulmonaires), ce qui confirme in vivo chez l’animal le rôle crucial (que laissaient supposer les études in vitro) de l’ACE2 dans l’infection par SARS-CoV (Kuba, Nat Med 2005 ; 11 :875-79 ). Dans ce modèle, les auteurs confirment chez des souris wild type que l’infection par SARS-CoV conduit à une sous-régulation très marquée de l’expression protéique de l’ACE2 dans le poumon. En outre, les auteurs ont montré que les antagonistes des récepteurs AT1 sont protecteurs chez des souris après injection de la protéine du spike de SARS-CoV (Kuba, Nat Med 2005).
En résumé, dans le SARS (-Cov1) il a été démontré sur des modèles expérimentaux in vivo que la liaison du virus à l’ACE2 entraine la downregulation de cette dernière, ce qui augmente les taux d’AngII et favorise les lésions pulmonaires (Kuba, Nat Med 2005 ; 11 :875-79 ; Gu et al, Am J Pathol 2007 ;170:1136–1147 ).
Données concernant la balance ACE/ACE2 au cours des infections pulmonaires diverses et par le SARS-CoV-2 chez l’homme
Les données chez l’homme sont nettement plus parcellaires mais semblent aller dans le même sens d’un rôle protecteur de la voie ACE2-Ang(1-7) et délétère de la voie ACE-AngII, cette dernière étant activée, au cours des pneumonies de différentes origines.
Ainsi une méta-analyse (Caldeira et al BMJ 2012, 345 :e4260) a mis en évidence une diminution du risque de survenue d’une pneumonie chez les patients traités par un IEC en comparaison à un placebo (19 études: OR 0.66, 95% CI 0.55 – 0.80). Cependant aucun effet protecteur n’était observé sous ARA2. Dans une étude rétrospective conduite chez 539 patients hospitalisés pour pneumonie virale (Texas), un traitement préalable par IEC était associé à une augmentation du risque combiné de mortalité ou d’intubation, mais ces mêmes risques étaient franchement diminués lorsque le traitement était poursuivi pendant l’ensemble de l’hospitalisation (Henry et al, Proc (Bayl Univ Med Cent) 2018;31(4):419-423). Bien entendu le caractère observationnel de ces données en limite l’interprétation puisque les biais potentiels sont majeurs.
Une étude chinoise récente a montré sur un faible nombre de patients que les taux d’Ang II sont augmentés au cours du COVID-19 (Liu et al, Science China Life Science 2020).
Quant aux études observationnelles récentes ayant rapporté les données de patients atteints du COVID-19 et traités ou non par bloqueur du SRA à l’entrée, elles vont également dans l’ensemble plutôt dans le sens d’un rôle neutre ou protecteur de ces traitements (voir plus haut partie « bloqueurs du SRA et prédisposition aux formes sévères de COVID-19 »)
Implications thérapeutiques potentielles
Dans l’hypothèse où l’activation de la voie Ang II lors de l’infection participe à la pathogénie du COVID-19, bloquer la voie de l’Ang II par les sartans serait donc au contraire protecteur et à recommander chez les patients.
D’où les essais en cours qui testent l’efficacité du losartan au cours du COVID-19 (au moins deux essais en cours aux Etats-Unis, à suivre… ClinicalTrials.gov Identifier: NCT04312009 ; ClinicalTrials.gov Identifier: NCT04311177)
→ Il y a donc au moins autant d’arguments théoriques pour instaurer des bloqueurs du SRA (favoriser le ratio Ang(1-7)/Ang II) que pour les arrêter (afin de ne pas risquer d’augmenter le nombre de récepteurs au virus).
L’ACE2 fait partie des gènes activés par l’interféron
Autre fait très important à propos des liens entre ACE2 et réponse inflammatoire de l’organisme infecté, il a été récemment rapporté dans la revue Cell que l’ACE2 fait partie des gènes stimulés par l’interféron α (Ziegler, Cell 2020 https://doi.org/10.1016/j.cell.2020.04.035). L’interféron sécrété suite à la pénétration intracellulaire du virus dans les tissus cibles pourrait alors enclencher une aggravation de l’infection en augmentant l’expression du récepteur membranaire du virus.
Forme circulante de l’ACE2 : quelles implications ?
Enfin l’ACE2 est également clivée par la métalloprotéinase ADAM17 et donc présente sous forme circulante dans le sérum où elle pourrait alors, au contraire, inactiver les particules virales circulantes [Batlle, Clin Sci 2020]. D’ailleurs, l’utilisation de l’ACE2 recombinante humaine soluble (APN01) est une piste thérapeutique chez les patients infectés par le COVID-19 (Clinicaltrials.gov #NCT04287686). Cette protéine recombinante pourrait aider à « neutraliser » la charge virale et/ou à restaurer la balance ACE2-Ang(1-7)/ACE-Ang II, qui pourrait jouer un rôle clé dans les lésion d’organes au cours du COVID-19 comme discuté plus haut.
Autant dire que le débat « bloqueurs du SRA et COVID-19, bon ou mauvais ?», est loin d’être tranché !
Possibilité d’une prédisposition génétique au COVID 19 ?
Est-ce que les polymorphismes de l’ACE et l’ACE2 modifient l’expression membranaire de l’ACE2 ou l’affinité du virus pour son récepteur, et donc la dangerosité d’une exposition au virus ? Impossible de le dire à l’heure actuelle, mais il faut trouver une explication à la raison pour laquelle certains malades font un syndrome de détresse respiratoire aigüe (SDRA) extrêmement sévère, tandis que d’autres ont une symptomatologie proche du rhume banal, ainsi qu’à la nette prédominance masculine dans les formes graves de la maladie… et ces polymorphismes sont une piste parmi d’autres.
L’ACE2 sur le chromosome X et comporte une centaine de polymorphismes, sans lien établi avec le COVID-19 à notre connaissance.
S’agissant de l’ACE, il existe des données potentiellement très pertinentes sur le lien entre polymorphismes et infection pulmonaire. Environ la moitié de la variance dans l’activité de l’ACE plasmatique est expliquée par le polymorphisme insertion/délétion (I/D) de l’intron 16 du gène de l’ACE, l’allèle D étant associé à une plus grande activité enzymatique de l’ACE. De manière très intéressante, il a été montré que ce polymorphisme de l’ACE est associé à la sévérité du SDRA (Marshall, AJRCCM 2002 ; 166 :646-50). Dans une étude conduite chez 96 patients présentant un SDRA en comparaison à diverses cohortes contrôles, les auteurs ont mis en évidence une plus grande prévalence du génotype DD chez les patients SDRA. De même, dans une étude conduite chez des patients atteints du SARS en 2003, il a été montré une fréquence plus élevée de l’allèle D de l’ACE dans le groupe hypoxémique en comparaison au groupe non hypoxémique (Itoyama et al, BBRC 2004 ; 323,1124-29). La fréquence de l’allèle D étant plus fréquente chez les patients atteints de pathologies cardiovasculaires (Baudin, Clin Chem Lab Med 2002 ; 40 :256-65), cela pourrait constituer encore une autre piste pour expliquer la prévalence des antécédents cardiovasculaires dans les formes graves du COVID-19.
De manière discordante, une étude récente a montré une corrélation inverse entre la prévalence de l’infection à COVID-19 et la fréquence de l’allèle D (Delangue et al, Clinica Chimica Acta 2020 ; 505 : 192–193). Selon ces auteurs, l’allèle D aurait au contraire un rôle protecteur, car il serait associé à une moindre expression de l’ACE2. Ces hypothèses nécessitent d’être étayées par des données plus solides au cours de l’infection à SARS-CoV-2
Utilisation des bloqueurs du SRA en réanimation
Bien entendu, chez le malade hospitalisé en réanimation, la question de laisser ou non en place le blocage du SRA dans ce contexte aigu fait intervenir des éléments de réflexion hémodynamiques tout autres que celui de l’arrêt préventif à l’échelle de la population qui est évoqué dans les récents éditoriaux sus-cités. Les réanimateurs connaissent parfaitement cette question, et en tout cas à court terme, le fait que ACE2 soit le récepteur du virus ne leur fera pas modifier leurs pratiques (le plus souvent, arrêt des bloqueurs du SRA en réanimation).
Les recommandations des différentes sociétés savantes qui en résumé disent de ne pas modifier notre pratique en l’absence d’éléments scientifiques solides paraissent donc tout à fait cohérentes y compris avec la vision spécifique du réanimateur, à la lumière des éléments actuels (qui peuvent évoluer rapidement).
Atteintes cardiaques au cours du COVID-19
Globalement, le profil d’expression d’ACE2 explique la diversité des atteintes d’organes au cours du COVID-19. Des atteintes cardiaques sont ainsi très fréquentes chez les patients infectés, ce qui est attendu au vu de la forte expression de l’ACE2 dans le cœur. Les atteintes cardiovasculaires observées au cours du COVID-19 incluent des myocardites, des troubles du rythme, des thromboses veineuses et embolies pulmonaires (pour revue, voir Driggin et al, JACC 2020, et Clerkin et al, Circulation 2020). Il semblerait que la tachycardie soit également un signe clinique fréquent chez les patients atteints y compris de formes bénignes, indépendamment de la fièvre (pas de publication claire à ce jour ni d’hypothèse mécanistique sur le sujet à notre connaissance). Bien entendu, les mécanismes lésionnels sont multiples et en partie indépendants d’un effet viral direct (orage cytokinique notamment).
Atteintes rénales au cours du COVID-19
Autre monitoring important chez les patients atteints du COVID19, qui découle directement du profil d’expression de l’ACE2, celui de la fonction rénale et de la protéinurie. La forte expression de l’ACE2 dans de nombreux types cellulaires du rein est bien connue et le virus a été détecté dans l’urine. La plus forte expression d’ACE2 dans le rein est retrouvée à la membrane apicale du tubule proximal et dans les podocytes (Soler et al, Curr Hypertens Rep 2008, 10: 410 – 414 ; Soler et al, Exp Physiol 2008, 93: 549– 553).
Une insuffisance rénale aigue et une protéinurie de fort débit ne sont pas rares dans le COVID-19, et sont décrites comme associées à un pronostic péjoratif (pour revue, voir Naicker et al, Kidney Int 2020 doi: 10.1016/j.kint.2020.03.001, voir également Perico et al, Nephron 2020 DOI: 10.1159/000507305).
A noter que malgré la forte expression de l’ACE2 dans le tubule proximal à ce jour il n’a pas été rapporté de syndrome de Fanconi au cours du COVID-19.
Une revue « rein et COVID » sera prochainement disponible sur le site de la Société de Physiologie.
COVID-19 et homéostasie potassique
Si la destruction/inactivation de l’ACE2 « détourne » le SRA en faveur de l’Ang II (Liu et al, Science China Life Science 2020), on peut attendre la survenue d’un hyperaldostéronisme avec fuite rénale de potassium au cours du COVID-19. Ce point a été rapporté sans investigation physiopathologique détaillée (Chen, in press, MedRxiv 2020): à suivre également…
EN RESUME :
L’ACE2 membranaire, exprimée non seulement dans le poumon, mais également dans le cœur, le rein et l’intestin, est le récepteur du SARS-CoV-2. Certains ont émis l’hypothèse que l’expression membranaire de l’ACE2 soit augmentée chez les patients traités par les bloqueurs du SRA, traitements qui pourraient le cas échéant prédisposer à l’infection par le COVID-19 ou à une forme plus sévère de la maladie. Des données observationnelles publiées ces derniers jours ne confortent pas l’hypothèse d’un rôle délétère de ces traitements vis-à-vis du COVID-19.
A contrario, le rôle potentiel de l’ACE2 au cours du COVID-19 est très ambivalent – « a doubleedged sword » comme la dénommait un commentaire récent de la revue Circulation (Wang et al, Circulation 2020) – et pourrait également être protecteur au cours de l’infection.
En effet, l’ACE2 est un homologue de la beaucoup plus célèbre ACE. L’ACE permet la synthèse de l’Ang II, qui via son récepteur AT1R a des effets vasoconstricteurs, pro-fibrosants, et globalement délétères dans plusieurs modèles de lésions pulmonaires. L’ACE2 a un rôle modulateur de l’ACE. Elle convertit l’Ang II en Ang(1-7), peptide vasodilatateur et anti-fibrosant, qui pourrait limiter la perméabilité capillaire et les lésions pulmonaires au cours du SDRA. L’inactivation de l’ACE2 par le virus pourrait jouer un rôle physiopathologique majeur au cours du COVID-19, en déviant la balance homéostatique ACE-Ang II/ACE2-Ang(1-7) vers la voie ACE-Ang II. Dans cette perspective, bloquer l’action de l’Ang II par exemple par des antagonistes du récepteur AT1 serait au contraire une piste thérapeutique intéressante.